
Molecular dynamics (MD) simulations are a robust tool in computational chemistry and materials science, and so they’re essential for studying chemical reactions, material properties, and biological interactions at a microscopic level. Nonetheless, their complexity and computational requirements often necessitate advanced techniques similar to machine learning interatomic potentials (MLIPs) to realize scalability, efficiency, and accuracy.
Integrating PyTorch-based MLIPs into the LAMMPS MD package through the ML-IAP-Kokkos interface enables fast and scalable simulation of atomic systems for chemistry and materials science research.
This interface was developed through a collaborative effort between NVIDIA, Los Alamos National Lab, and Sandia National Lab scientists. It was designed to simplify the technique of interfacing community models for external developers, allowing them to seamlessly connect MLIPs with LAMMPS enabling scalable MD simulations.
The ML-IAP-Kokkos interface supports message-passing MLIP models, utilizing LAMMPS’s built-in communication capabilities to facilitate efficient data transfer between GPUs—something that’s crucial for enabling large-scale simulations that leverage the computational power of multiple GPUs. The interface uses Cython to bridge Python and C++/Kokkos LAMMPS, ensuring end-to-end GPU acceleration, optimizing the general simulation workflow.
We’ll walk you thru the right way to connect your personal PyTorch models for scalable LAMMPS simulation, step-by-step. Let’s start.
- Experience with LAMMPS or other MD simulation tools.
- Experience using Python, familiarity with PyTorch and machine learning models (Cython a plus).
- Optional: for added acceleration, cuEquivariance supported models and NVIDIA GPUs.
Constructing your personal ML-IAP-Kokkos Interface
Step 1: Organising the environment
Ensure you may have all obligatory software installed:
- LAMMPS built with Kokkos, MPI, and ML-IAP
- Python with PyTorch
- Trained PyTorch MLIP model (optionally with cuEquivariance support)
Step 2: Install LAMMPS with ML-IAP-Kokkos/Python support
Download and install LAMMPS (September 2025 release or later) with Kokkos, MPI, ML-IAP and Python support.
Installing LAMMPS with Python support also installs the lammps module in your Python environment.
For convenience, we’ve prepared a container with a precompiled LAMMPS.
Step 3: Develop ML-IAP-Kokkos interface for preferred model
When using the ML-IAP-Kokkos interface, LAMMPS will call the Python interpreter throughout the simulation, executing your native Python model and providing all Python features without having to compile your model. This requires a working Python environment with access to your model’s classes.
To attach your model with LAMMPS, you should implement the abstract class MLIAPUnified from LAMMPS as defined in `mliap_unified_abc.py`. Specifically, you should make the compute_forces function infer pairwise forces and energies using the information passed to it from LAMMPS (the category will even require compute_gradients and compute_descriptors functions, but these could be empty as they may not be utilized in this setup).
Starting with a straightforward example to know the information that LAMMPS passes to your class, we create a category that prints some information:
from lammps.mliap.mliap_unified_abc import MLIAPUnified
import torch
class MLIAPMod(MLIAPUnified):
def __init__(
self,
element_types = None,
):
super().__init__()
self.ndescriptors = 1
self.element_types = element_types
self.rcutfac = 1.0 # Half of radial cutoff
self.nparams = 1
def compute_forces(self, data):
print(f"Total atoms: {data.ntotal}, Local atoms: {data.nlocal}")
print(f"Atom indices: {data.iatoms}, Atom types: {data.elems}")
print(f"Neighbor pairs: {data.npairs}")
print(f"Pair indices and displacement vectors: ")
print("n".join([f" ({i}, {j}), {r}" for i,j,r in zip(data.pair_i, data.pair_j, data.rij)]))
Note that the __init__ function specifies the element types our model can handle and (half) the neighbor list’s cutoff radius. These parameters are required by LAMMPS in model setup.
We save our model object with the PyTorch save function:
mymodel = MLIAPMod(["H", "C", "O"])
torch.save(mymodel, "my_model.pt")
This creates the file my_model.pt, a “pickled” Python object, which LAMMPS can load. In an actual scenario this may contain the weights of our model but here it’s a serialization of our class. (In the most recent versions of PyTorch, loading a category is disabled by default as a security protection because the class incorporates arbitrary Python code. Setting the environment variable TORCH_FORCE_NO_WEIGHTS_ONLY_LOAD=1 allows loading the category. Use only trusted *.pt files).
For testing, we’ll define a system of a single CO2 molecule within the file sample.pos:
#The mandatory first line is a comment and skipped!
3 atoms
2 atom types
0.0 100.0 xlo xhi
0.0 100.0 ylo yhi
0.0 100.0 zlo zhi
Masses
1 12.0
2 16.0
Atoms
1 2 1.0 10.0 10.0
2 1 2.0 10.0 10.0
3 1 -0.1 10.0 10.0
The molecule consists of 1 carbon atom and two oxygen atoms arranged linearly along the X-axis, with the carbon positioned at x = 1.0 Å and the oxygen atoms at x = -0.1 Å and x = 2.0 Å, respectively. This minimal configuration allows us to obviously observe how LAMMPS handles local versus ghost atoms, pair interactions, and periodic boundary conditions—key elements for understanding the information flow within the interface.
Figure 1 illustrates the atomic positions and their relationship to the simulation cell boundaries.
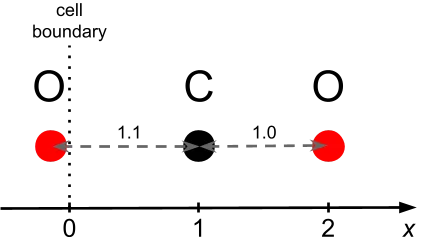
The next LAMMPS input sample.in loads the position file, loads our potential, and runs a single MD step:
units metal
atom_style atomic
atom_modify map array
boundary p p p
read_data sample.pos
# Load the ML-IAP model
pair_style mliap unified my_model.pt 0
pair_coeff * * C O
dump dump0 all custom 1 output.xyz id type x y z fx fy fz
run 0
Using the pair style mliap, the “unified” keyword directs LAMMPS to make use of the Python interface, followed by the model file to load, and at last the worth 0, which affects the neighbor list (see documentation for details).
We specify the subset of elements we’re fascinated by (C and O) and their mapping to the indices within the positions file.
We run LAMMPS with Kokkos on one GPU with the command:
lmp -k on g 1 -sf kk -pk kokkos newton on neigh half -in sample.in
And take a look at this relevant a part of the output:
Total atoms: 6, Local atoms: 3
Atom indices: [0 1 2], Atom types: [2 1 1 2 1 1]
Neighbor pairs: 4
Pair indices and displacement vectors:
(0, 5), [-1.1 0. 0. ]
(0, 1), [1. 0. 0.]
(1, 0), [-1. 0. 0.]
(2, 3), [1.1 0. 0. ]
Comparing this with our class, we understand the information structure:
- Atoms are divided into two categories: physical atoms, starting from
[0, nlocal), which need to be updated; and ghost atoms ranging from[nlocal, ntotal), which are either periodic copies or physical atoms from another processor/GPU. - There are three local atoms (
nlocal) and three ghost atoms for a total of six (ntotal). iatomslists the indices of local atoms (the first nlocal).elemslists the atomic species of all atoms.npairsis the number of pairs within the cutoff that involve at least one local atom.
Note that pairs (i, j) and (j, i) are reported separately: here we have four pairs corresponding to the two (C, O) bonds in either direction.
We can access the (i, j) indices of each pair and the relative displacement via pair_i, pair_j, and rij.
Note that indices greater than or equal to nlocal refer to ghost atoms (e.g. 3 is the periodic ghost of 0, 5 is the periodic ghost of 2).
To implement a simple potential, imagine a model that computes some “node features” on local atoms and updates the energy based on neighbor features and bond lengths, a typical setup for MLIPs. For simplicity, take all features to be 1 and the radial function to be the atomic distance (in an actual implementation, these would be more complex objects computed by our model).
A (partial) implementation of this potential might be:
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Message passing step (j → i)
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
Some details to note on this code:
- All quantities passed between the ML-IAP and LAMMPS are fp64 and on device, although type-casting and host<->device movement is permitted within the model.
- Total energy is updated directly in our model.
- We compute forces through the autograd function with respect to rij, i.e. forces are computed “per bond” and never “per atom.” Potentials that compute atomic forces directly should be recast in this type.
We update the forces in LAMMPS through the function update_pair_forces_gpu, passing the fp64 GPU tensor of shape (npairs, 3).
We test this code on our small CO2 molecule.
For reference, we compute energies and forces by hand with only two pairs and two distances (1 and 1.1), the whole energy ought to be 4.2, with each pair contributing 1 to the force, yielding a force of two in opposite directions for the carbons, with 0 force on the oxygen.
Nonetheless, running this code we obtain a complete energy of two (as seen in stdout) and force 2 (as seen in output.xyz) on one carbon and the oxygen. It’s clear that the issue is the presence of ghost atoms; their features will not be updated on the true atoms in order that only the (0, 1) bond contributes to the whole energy and forces.
On this simplified case, we could initialize the ghosts to 1, but in an actual scenario, this may be a more complex function of the environment. In that case, we use the message passing hooks built into ML-IAP-Kokkos to update the ghost features to the worth of their corresponding real counterparts. This takes care of ghost particles coming from periodic boundary conditions and people residing on a wholly different GPU.
Two routines are provided to assist this initialization process: forward_exchange, which sets ghost atoms from their physical counterparts; and reverse_exchange, which sums contributions made to ghost atoms back to their physical counterparts.
Step 4: Implementing message passing support for ML-IAP-Kokkos
Updating our model to make use of the message passing step (see code below), we call forward_exchange, which takes two tensors of shape (ntotal, feat_size) and sets features[0,nlocal) to new_features[0,ntotal), handling local copying and MPI messaging. The feat_size, 1 here, is the vector width of the feature. This function creates a copy of features with ghost values set from their physical counterparts.
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Update of the features
new_features = torch.empty_like(features)
data.forward_exchange(features, new_features, 1)
features = new_features
# Local message passing step
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
Running this code again, we now obtain the proper energy and forces. Nonetheless, on multiple GPUs, the force calculation wouldn’t work accurately because the gradients will not be computed across different devices.
To repair this, we want to define and register the gradient of LAMMPS_MP.forward() with PyTorch. The gradient of forward_exchange is provided via reverse_exchange. After calling reverse_exchange, gradients are confined to real atoms and zeroed on all ghost atoms.
Here is an easy implementation of the whole message passing:
import torch
class LAMMPS_MP(torch.autograd.Function):
@staticmethod
def forward(ctx, *args):
feats, data = args # unpack
ctx.vec_len = feats.shape[-1]
ctx.data = data
out = torch.empty_like(feats)
data.forward_exchange(feats, out, ctx.vec_len)
return out
@staticmethod
def backward(ctx, *grad_outputs):
(grad,) = grad_outputs # unpack
gout = torch.empty_like(grad)
ctx.data.reverse_exchange(grad, gout, ctx.vec_len)
return gout, None
With this latest class, the ultimate version of our plugin becomes:
def compute_forces(self, data):
rij = torch.as_tensor(data.rij)
rij.requires_grad_()
features = torch.zeros((data.ntotal,1)).cuda()
for i in range(data.nlocal):
features[i,0] = 1.0
Ei = torch.zeros(data.nlocal).cuda()
# Update of the features
features = LAMMPS_MP().apply(features, data)
# Local message passing step
for r,i,j in zip(rij, torch.as_tensor(data.pair_i), torch.as_tensor(data.pair_j)):
Ei[i] += features[j,0] * torch.norm(r)
# Compute total energy and forces
Etotal = torch.sum(Ei)
Fij = torch.autograd.grad(Etotal, rij)[0]
# Copying total energy and forces
data.energy = Etotal
data.update_pair_forces_gpu(Fij)
We now have access to proper multi-GPU message passing that accurately supports autograd.
This same structure should work independently of message passing MLIP models.
Calling LAMMPS_MP before any message passing ensures that features remain synchronized and the model works accurately. Optimization may exist, nonetheless, where some communication steps could also be skipped. Since LAMMPS collects ghosts before calling the ML-IAP and since LAMMPS propagates forces on ghosts to other ranks afterwards, the primary forward exchange and last reverse exchange in a multi-layer MLIP can often be skipped.
As mentioned, the force calculation requires the computation of energy gradients with respect to bonds. Moreover, passing all terms to LAMMPS allows LAMMPS to tally the contributions for the computation of stresses; stresses don’t should be computed by the plugin.
To exhibit the performance improvements that the message passing hooks enable, we benchmarked LAMMPS running with HIPPYNN (Chigaev 2023) models in multiple series of weak scaling tests on one to 512 NVIDIA H100 GPUs with models comprised of h=1 to h=4 interaction layers within the MLIP with and without the communication hooks. We use a straightforward lattice of aluminum atoms as a performance test, keeping the variety of atoms to roughly 203 atoms per GPU.
The models were trained on the dataset from the paper Automated discovery of a sturdy interatomic potential for aluminum following the example multi-interaction layer training script in HIPPYNN. For simulation correctness, where r is the interaction radius of 1 MLIAP layer, we use a ghost cutoff length r+epsilon with the hooks enabled and hr+epsilon with ghost-ghost pairs enabled within the neighbor list when the hooks are disabled.
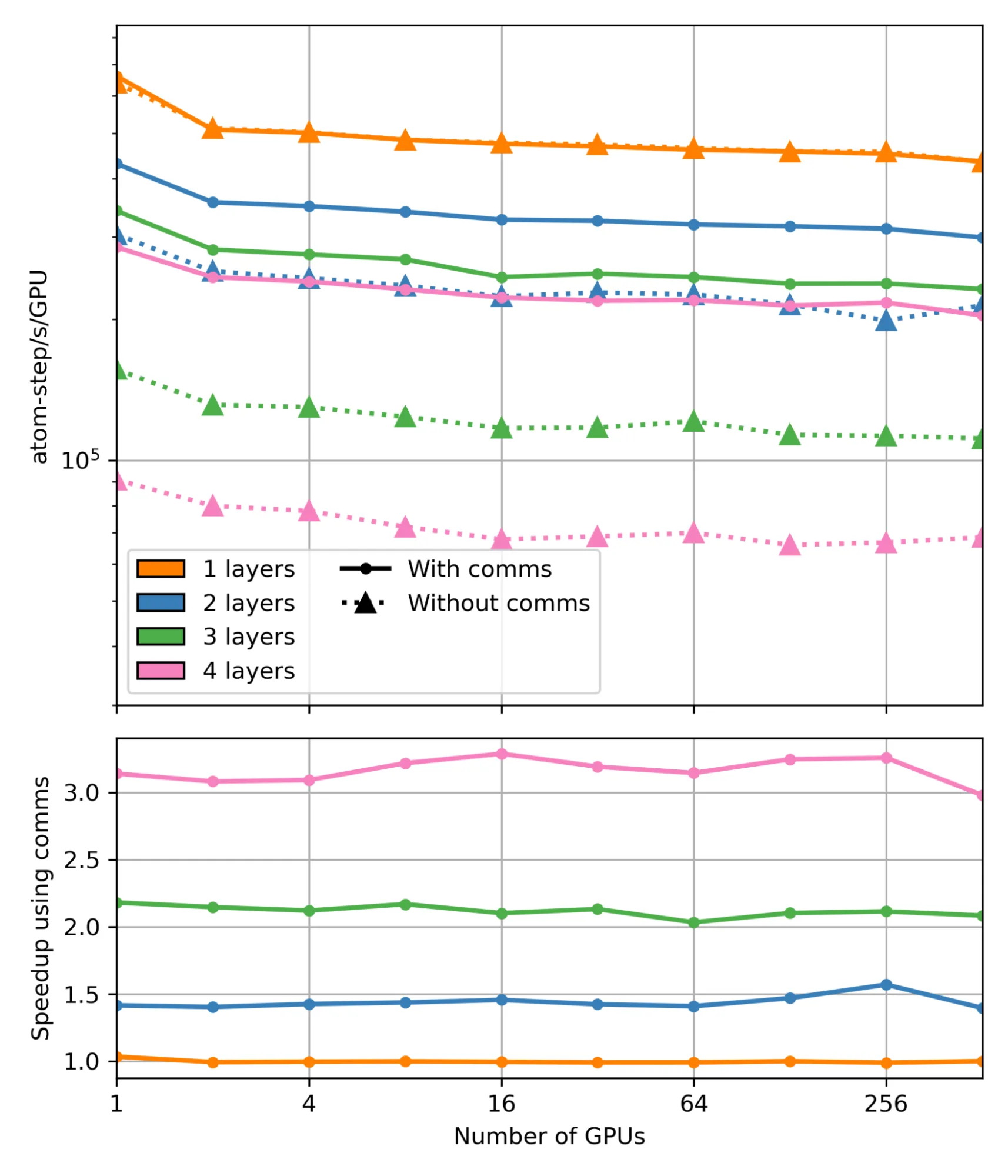
The speedup from using the communication hooks shown within the figure above is directly attributable to the reduction in ghost atoms. The proportion of real atoms to total atoms (real plus ghost) with one, two, three, and 4 layers of ghost atoms is 54%, 38%, 26%, and 18%, respectively. With hooks enabled, we will use one radius of ghosts and thus 54% real atoms for all cases. This results in a 1.4x, 2.1x, and 3x reduction in total atoms for 2, three, and 4 layers, respectively (for one layer, the hooks are non-operative), directly corresponding to the observed speedups.
Comparing results using MACE integration
As one further demonstration, we benchmark LAMMPS simulations using the favored MLIP MACE comparing the unique MACE pairstyle in LAMMPS, which utilizes libtorch in C++ to load model weights, to a cuEquivariance-based MACE plugin using the MLIAP-Kokkos interface. cuEquivariance is an NVIDIA Python library for accelerating geometric neural networks.
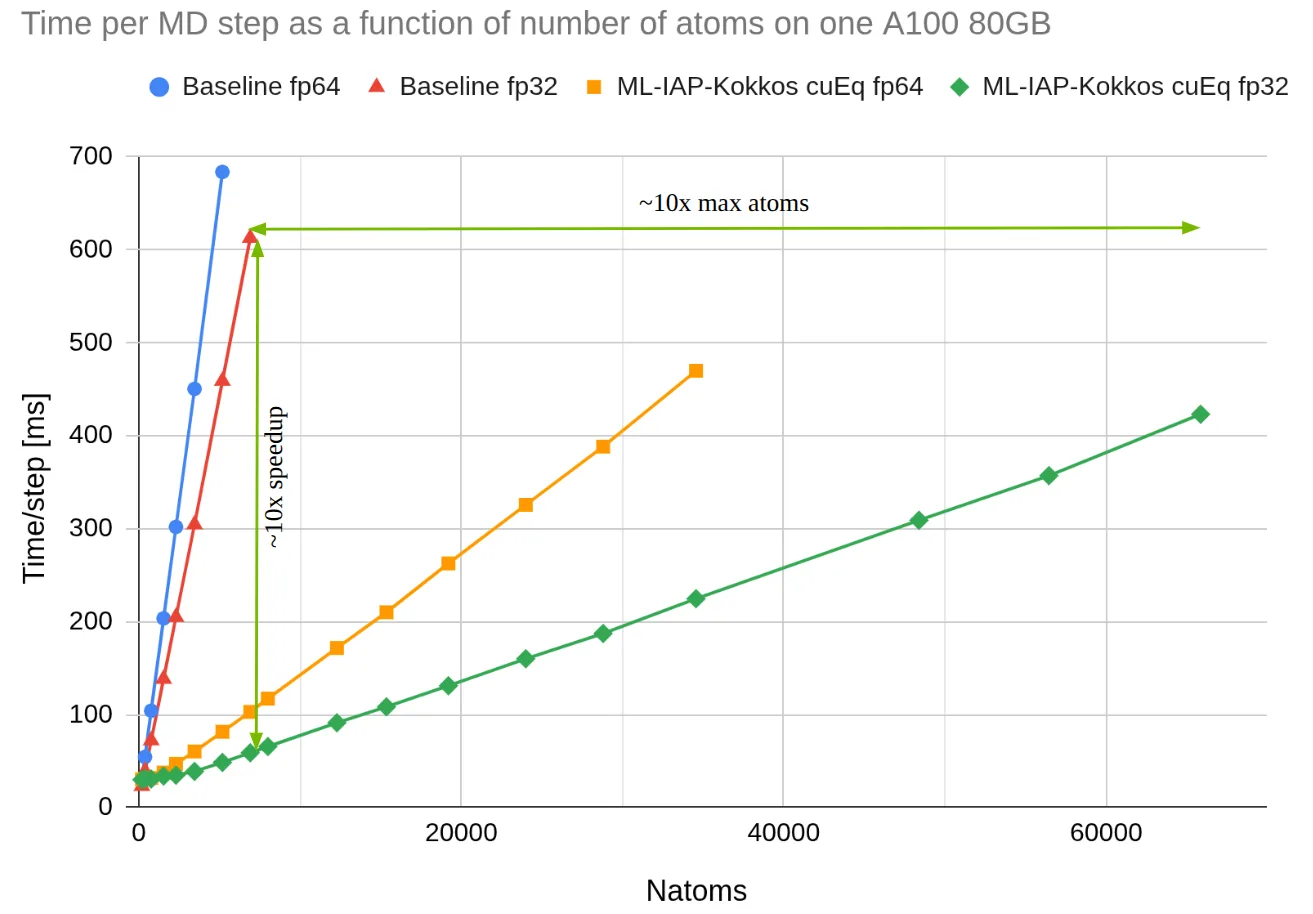
For this benchmark, we simulate increasingly larger boxes of water until GPU memory is exhausted using the “medium” version of the MACE-OFF23 potential on a single A100 GPU with 80GB of memory with fp32 and fp64 models. The speed of the unique MACE pairstyle is shown for comparison. The brand new plugin is much faster and more memory efficient. This speedup is attributable to model acceleration with cuEquivariance and the message passing improvements within the MLIAP-Kokkos interface. CuEquivariance accelerated MLIPs could be seamlessly utilized in LAMMPS when passing through the ML-IAP-Kokkos interface.
Conclusion
The LAMMPS ML-IAP-Kokkos interface is currently crucial tool enabling multi-GPU, multi-node MD simulations using MLIPs. It allows the simulation of extremely large systems with high efficiency, bridging the gap between modern ML-based force fields and high-performance computing infrastructures.
By integrating PyTorch-based MLIPs with LAMMPS via the ML-IAP-Kokkos interface, developers can achieve scalable and efficient MD simulations. This tutorial outlines the steps obligatory to establish and execute simulations and highlights the advantages of GPU acceleration and message-passing capabilities in LAMMPS. The benchmark results further emphasize the interface’s potential for large-scale simulations, making it a helpful tool for researchers and developers in the sector.
Able to speed up your simulations? Try the tutorial today and share your results with the community. Subscribe to NVIDIA news and follow NVIDIA AI on LinkedIn, X, Discord, and YouTube.